兩電子氧化在合成和催化中無處不在,并起着舉足輕重的作用。對于過渡金屬和锕系元素來說,兩電子氧化常發生在單個金屬中心,如氧化加成反應和氧原子轉移反應等(圖1a)。然而,由于缺乏可變氧化态,稀土元素的氧化反應局限于單電子過程,如常用的單電子還原劑SmI2和單電子氧化劑(NH4)2[Ce(NO3)6],而兩電子過程則需要兩個或以上金屬中心的參與(圖1b)。近年來,稀土元素非尋常氧化态化學的發展為實現單一稀土元素中心的兩電子過程提供了可能。但是,非正三價稀土離子的不穩定性以及不同價态稀土離子間的巨大差異阻礙了這一目标的實現。近日,beat365官方网站黃聞亮課題組利用三腳架型三胺基芳烴配體成功穩定了铈的三種氧化态(+2到+4價),并首次實現了從Ce(II)到Ce(IV)的兩電子氧化(圖1c)。該成果以“Two-electron oxidations at a single cerium center”為題,于2023年9月22日在《美國化學會志》(Journal of the American Chemical Society)上在線發表,并被選為封面文章。

圖1:稀土元素與過渡金屬和锕系元素氧化還原化學的比較
該課題組近年來基于“f區金屬–芳烴協同作用”的概念,發展了一類三腳架型三胺基芳烴配體并将其引入f區金屬配位化學(圖2a)。最近,利用金剛烷基取代的配體((AdTPBN3)3‒)來穩定鈾的五種氧化态(+2到+6價),并證實了底座芳環能夠起到同時穩定低價和高價鈾離子的作用(圖2b)。在此工作中,作者利用該配體穩定铈的三種氧化态,+2到+4價,并首次實現了從Ce(II)到Ce(IV)的兩電子氧化反應(圖2c)。

圖2:三腳架型三胺基芳烴配體支撐的f區金屬配位化學
從配體鉀鹽K3(AdTPBN3)和三價铈前驅體CeX3(THF)4 (X = Br or I)出發,通過鹽複分解反應可得三價铈的配合物(AdTPBN3)Ce(III) (1)。電化學測試結果顯示,1在‒2.68 V (vs. Fc+/Fc)處有一個可逆的還原峰,而在‒0.26 V (vs. Fc+/Fc)則有一個不可逆的氧化峰,表明1的化學還原和氧化是可行的。用KC8/crypt(穴醚222)還原1可以得到二價铈的配合物[K(crypt)][(AdTPBN3)Ce(II)] (K[2])(圖3a)。核磁氫譜顯示,2中底座芳烴的氫位于極高場‒178 ppm,表明铈與底座芳烴存在較強的軌道相互作用。盡管1與一些常用氧化劑無法得到明确的結果,但利用TeCl4氧化後再加Me3SnF處理的方法,可以得到穩定的四價铈的氟化物(AdTPBN3)Ce(IV)F (4);此外,1與KOtBu反應後再通入氧氣可以得到四價铈的叔丁氧配合物(AdTPBN3)Ce(IV)OtBu (5)(圖3b)。這些結果表明(AdTPBN3)能夠穩定铈的三種氧化态(+2到+4價),為進一步嘗試從二價铈到四價铈的直接兩電子氧化打下了基礎。K[2]與吡啶氧化物或3,5-雙(三氟甲基)苯基疊氮(N3ArCF3)反應,能夠得到四價铈的末端氧配合物(AdTPBN3)Ce(IV)O (K[7])或末端亞胺配合物(AdTPBN3)Ce(IV)NArCF3 (K[8])(圖3c)。這是首個稀土元素的單中心兩電子氧化反應。

圖3:铈配合物的合成及其氧化還原化學:a 三價铈還原為二價铈;b 三價铈的單電子氧化反應;c 二價铈的兩電子氧化反應
作者對铈配合物進行了全面的譜學、結構和磁性表征,包括核磁共振波譜、紫外‒可見‒近紅外吸收光譜、紅外光譜、單晶X射線衍射、超導量子幹涉儀磁學(SQUID)和電子順磁共振(EPR)的表征,來探究其電子結構。二價铈配合物K[2]的變溫磁化率測試表明,在低溫下其磁矩趨近于零,符合4f2電子構型對應的3H4非磁基态;但在室溫下其磁矩顯著低于“自由4f2”的三價镨離子,表明4f軌道可能參與成鍵。低溫EPR測試顯示,K[2]的固體樣品沒有EPR信号,而凍溶液樣品僅有微弱的可歸屬于溶劑化電子的信号,排除了K[2]為三價铈離子和芳烴自由基陰離子的可能,進一步支持K[2]為4f2電子構型的二價铈配合物。
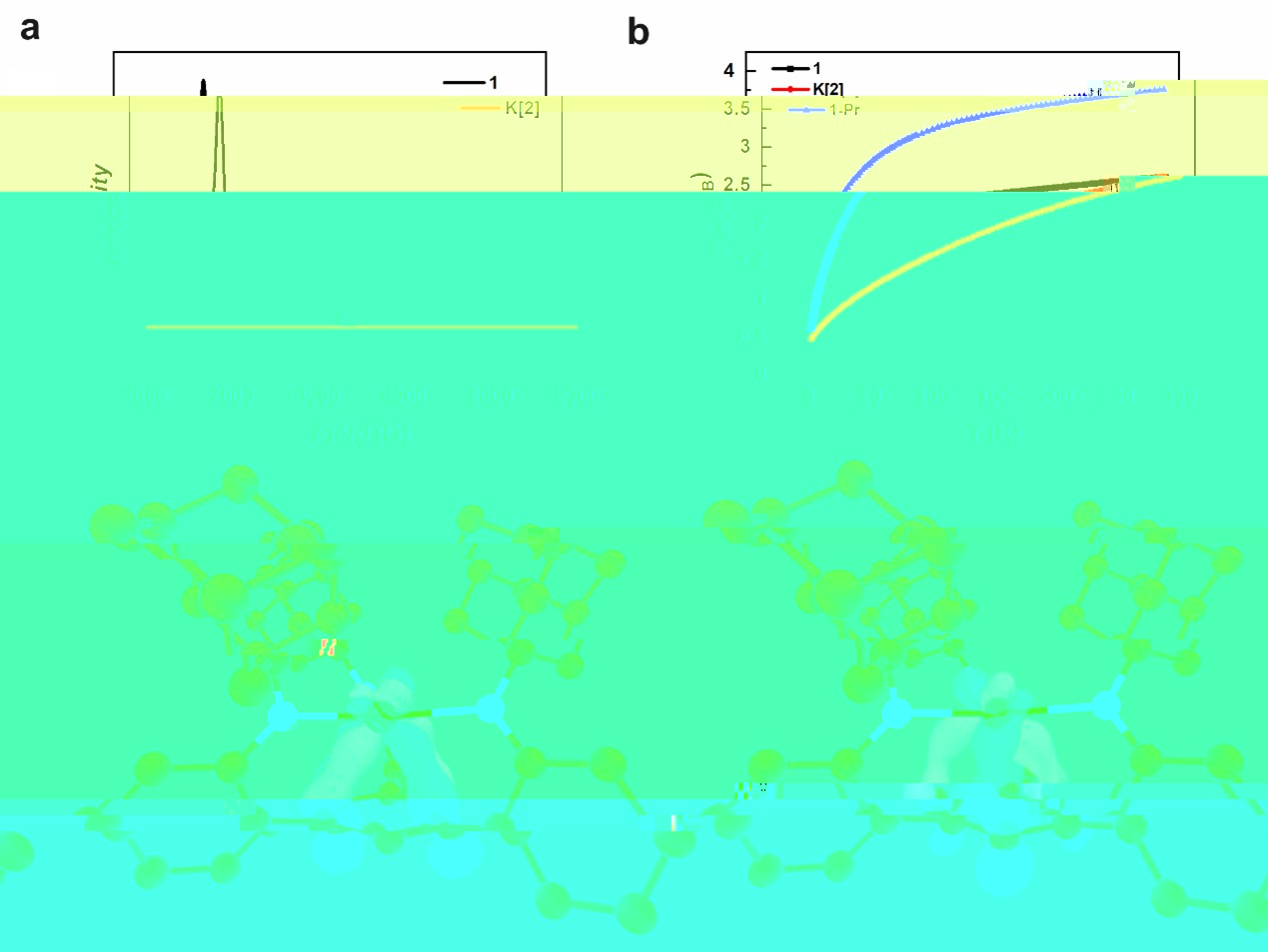
圖4:二價铈配合物K[2]的電子結構:a1和K[2]的固體樣品在10 K下的EPR譜;b1,K[2]和1-Pr在1kOe條件下的變溫磁化率數據;c K[2]的SOMO和SOMO‒1軌道
為了進一步探究K[2],K[7]和K[8]的電子結構和成鍵性質,作者進行了基于密度泛函理論(DFT)的計算研究。DFT計算結果顯示,[2]‒的SOMO和SOMO‒1軌道為近簡并的铈4f軌道和底座芳烴π*軌道的δ相互作用,且铈的4f軌道占比要高于底座芳烴2p軌道的占比。這一結果表明對二價铈配合物電子結構的最恰當表示是通過δ反饋鍵穩定的4f2電子構型,與文獻報道的大多數非傳統二價稀土離子4fn5d1的電子構型迥異。K[2]特殊的4f2電子構型與配合物結構有關:在近似C3v的配位場下,配體底座芳烴的π給電子性大幅擡高了dσ軌道的能量,而dπ和dδ軌道則參與了與配體胺基氮原子的成鍵,因此铈的5d軌道能量過高無法填充電子;另一方面,還原後铈與底座芳烴的距離顯著縮短,在小幅擡高fσ軌道能量的同時,通過铈與底座芳烴的δ成鍵作用大幅降低了fδ軌道的能量(圖5a)。這一系列因素最終促使二價铈離子采用4f2的電子構型。此外,作者還對四價铈的末端氧配合物K[7]和末端亞胺配合物K[8]的铈‒氧和铈‒氮多重鍵性質進行了自然局域分子軌道(NLMO)的分析。分析表明,在铈‒氧三重鍵中,兩個近簡并的π鍵能量顯著低于σ鍵,這在多重鍵中并不常見,之前僅見于鈾酰等少數例子中,凸顯了铈‒氧多重鍵的強度。與之相反,在铈‒氮多重鍵中,σ鍵能量遠低于π鍵,而且計算得到的Wiberg鍵級1.23也顯著低于铈‒氧鍵的1.58。

圖5:a還原前後價層軌道能量和電子排布的變化;b四價铈末端氧配合物的铈氧π4σ2三重鍵
綜上,該項工作合成了一系列三腳架型三胺基芳烴配體支撐的铈(II‒IV)配合物,并首次實現了稀土元素的單中心兩電子氧化。實驗表征和理論計算共同表明,二價铈配合物的電子結構為δ反饋作用穩定的4f2電子構型。此外,成鍵分析揭示了四價铈末端氧和末端亞胺配合物的铈‒氧和铈‒氮鍵多重鍵的特征。這些研究成果開啟了稀土氧化還原化學的新篇章,也展示了“金屬‒芳烴協同作用”這一策略在穩定多種氧化态和構建金屬多重鍵方面的廣闊前景。
該論文的第一作者為beat365官方网站博士研究生王怡,博士研究生梁傑鋒、鄧翀、孫榮、付鵬翔為該工作做出了貢獻,王炳武研究員和高松教授為共同作者,黃聞亮研究員為通訊作者。該研究得到了國家自然科學基金、國家重點研發計劃、beat365、北京分子科學國家研究中心等的資助,并得到了beat365分析測試中心、beat365高性能計算平台等的支持。
原文鍊接:https://doi.org/10.1021/jacs.3c06613
排版:高楊
審核:李玲,彭海琳

 北大化學微信
北大化學微信