


Unusual Electronic Structures of an Electron Transfer Series of [Cr(μ-η1:η1-N2)Cr]0/1+/2+
Gao-Xiang Wang,* Chunxiao Shan, Wang Chen, BotaoWu, Peng Zhang, Junnian Wei,* Zhenfeng Xi, Shengfa Ye*
Angew. Chem. Int. Ed., 2024. DOI: 10.1002/anie.202315386.
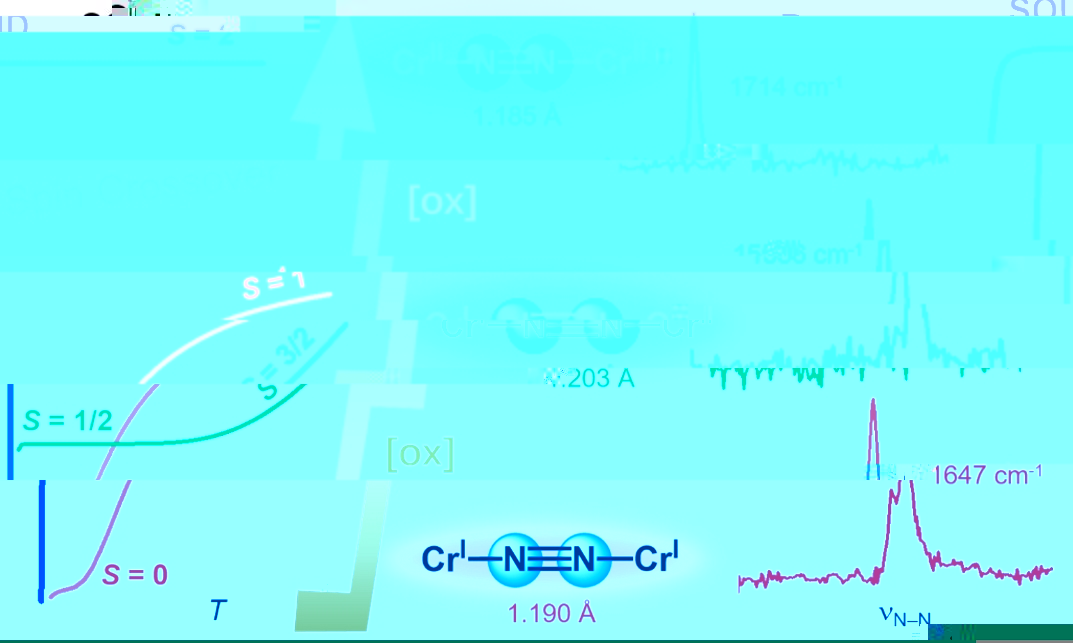
In dinitrogen (N2) fixation chemistry, bimetallic end-on bridging N2 complexes M(μ-η1:η1-N2)M can split N2 into terminal nitrides and hence attract great attention. To date, only 4d and 5d transition complexes, but none of 3d counterparts, could realize such a transformation. Likewise, complexes {[Cp*Cr(dmpe)]2(μ-N2)}0/1+/2+ (1-3) are incapable to cleave N2, in contrast to their Mo congeners. Remarkably, cross this series the N-N bond length of the N2 ligand and the N-N stretching frequency exhibit unprecedented nonmonotonic variations, and complexes 1 and 2 in both solid and solution states display rare thermally activated ligand-mediated two-center spin transitions (STs), distinct from discrete dinuclear spin crossovers. In-depth analyses using wave function based ab initio calculations reveal that the Cr-N2-Cr bonding in complexes 1-3 is distinguished by strong multireference character and cannot be described by solely one electron configuration or Lewis structure, and that all intriguing spectroscopic observations originate in their sophisticate multireference electronic structures. More critical is that such multireference bonding of complexes 1-3 is at least a key factor that contributes to their kinetic inertness toward splitting N2. The mechanistic understanding is then used to rationalize the disparate reactivity of related 3d M(μ-η1:η1-N2)M complexes compared to their 4d and 5d analogs.
雙核金屬端基橋式氮氣配合物M(μ-η1:η1-N2)M是氮氣活化與轉化過程中的關鍵中間體之一。不同于單核金屬氮氣配合物通過多步質子耦合的電子轉移反應(M-N2 → M-N=N-H → M=N-NH2 → M≡N)逐步斷裂氮氮鍵的活化模式,雙核金屬氮氣配合物可以直接斷裂氮氮三鍵,生成金屬氮化物(M(μ-η1:η1-N2)M → 2 M≡N)。目前,這類氮氣撕裂反應的報道主要集中于4d和5d金屬(Mo, W, Nb, Ta, Re等),3d金屬未見報道。在均相氮氣活化領域,“失敗”往往才是主旋律,相較于少數“成功”案例,探明3d金屬雙核氮氣配合物不能斷裂氮氣的内在原因更為關鍵。據此,課題組與大連化物所葉生發團隊合作,對雙核鉻橋式氮氣配合物的電子結構提出了新的見解,并闡釋了其不能發生氮氣撕裂反應的本質原因。
我們利用SQUID,EPR,Evans磁化率,Raman,UV-vis等一系列光譜學表征手段結合高精度的從頭算方法,開展了一系列雙核鉻氮氣配合物{[Cp*Cr(dmpe)]2(μ-N2)}0/1+/2+電子結構的系統性研究。實驗結果表明雙核鉻氮氣配合物在固态和溶液态下均存在自旋交叉(spin crossover)現象。其中,{[Cp*Cr(dmpe)]2(μ-N2)}0出現S = 0 到S = 1的自旋交叉,{[Cp*Cr(dmpe)]2(μ-N2)}1+出現S = 1/2 到S = 3/2的自旋交叉。這類雙核金屬配合物的自旋交叉現象極為罕見,自旋态的轉變并不是單個過渡金屬中心的行為,而是Cr-N2-Cr單元整體自旋态的變化。多光譜學表征結果說明這些鉻氮氣配合物具有很近的激發态,其電子結構具有多組态性質。CASSCF/NEVPT2的計算結果也進一步證實了雙核鉻氮氣配合物的多組态電子結構特征。
我們認為,正是由于這種多組态的金屬氮氣鍵合模式,“稀釋”了能夠發生氮氣撕裂的有效電子組态((1πu)4(2πg)4(3πu)2),因而氮氮鍵的裂解反應不能順利發生,即該反應本身存在着較大的動力學阻礙,不僅正向反應難以進行,逆向過程同樣無法發生。此外,這種多組态的性質也體現在随着逐步單電子氧化,雙核鉻氮氣配合物氮氮鍵鍵長(1.190(3) Å → 1.203(4) Å → 1.184(4) Å)和氮氮鍵伸縮振動頻率(1647 cm-1 → 1556 cm-1 → 1714 cm-1)的不規則變化上。幾何結構的變化是電子結構發生改變的直接體現,作者因為在晶體結構上注意到了這一細微的鍵長變化,才決定利用各種光譜學表征手段來闡明其背後的電子結構特征。這一成果近期發表在Angewandte Chemie International Edition上,文章的第一作者是beat365博士後王高翔。